In lab known for its quality data collection, high-speed video style, writing the weekly blog post can be a bit of a challenge for the local code monkey. That’s right, no videos today. But lucky for me, even this group can still make good use of publicly available data. Like the other day, when one of our newer graduate students wanted to do a certain degree of data-mining from information available from FishBASE. Now I’m sure there are plenty of reasons to grumble about it, but there’s quite an impressive bit of information available on FishBASE, most of it decently referenced if by no means comprehensive. And getting that kind of information quickly and easily is rapidly becoming a skill every researcher needs. So here’s a quick tutorial of the tools to do this.
Okay, so what are we talking about? Let’s start with an example fishbase page on Nile tilapia:
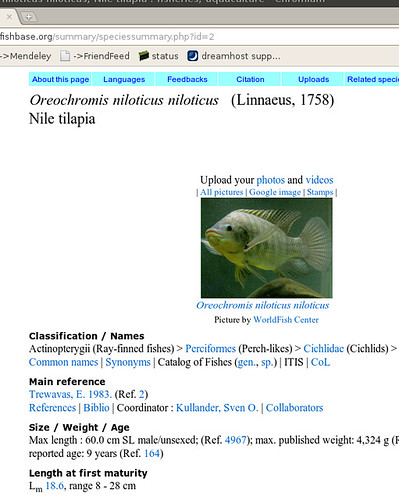
Okay, so there’s a bunch of information we could start copying down, then go onto the next fish. Sounds pretty tedious for 30,000 species… Now we can get an html copy of this page, but that’s got all this messy formatting we’ll have to dispense with. Luckily, we scroll down a little farther and we see the magic words:
Download XML
The summary page takes us to just what we were looking for:
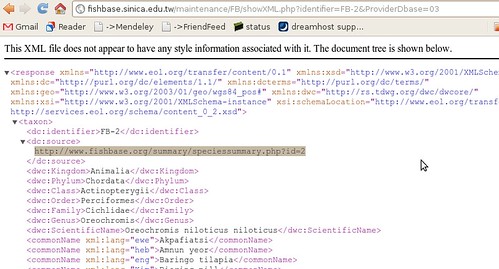
To the human eye this might not look very useful, but to a computer, it’s just what we wanted. ((Well, not acutally. A RESTful interface to this data would be better, where we could query by the different categories, finding all fish of a certain family or diet, but we’ll manage just fine from here, just might be a bit slower)) It’s XML – eXtensible Mark-up Language: meaning it’s all the stuff on the previous page, marked up with all these helpful tags so that a computer can make sense of the document. ((Actually, not everything on the html page. Between the broken links, there’s tons of information like the life history tool, diet data references, etc that somehow haven’t made it into the XML summary sheet.))
To process this, we’ll fire up our favorite language and start scripting. To access the internet from R we’ll use a the command-line url browser, curl, available in the RCurl package. We’ll process XML with the XML package, so let’s load that too. Install these from CRAN if you don’t have them:
[source lang=”r”] require(XML)require(RCurl)
[/source]
Click on that XML link to the summary page, and note how the url is assembled: http://fishbase.sinica.edu.tw/maintenance/FB/showXML.php?identifier=FB-2&ProviderDbase=03
The thing to note here is what followers the php?. There’s something called an identifier, which is set equal to the value FB-2. 2 is the id number of Nile tilapia. Change that to 3 (leave the rest the same) and you get African perch.
We can grab the content of this page in R and parse the XML using these two packages:
[source lang=”r”] fish.id <- 2url <- paste("http://fishbase.sinica.edu.tw/",
"maintenance/FB/showXML.php?identifier=FB-",
fish.id, "&ProviderDbase=03", sep="")
tt <- getURLContent(url, followlocation=TRUE)
doc <- xmlParse(tt)
[/source]
We create the url with a given fish id. Then the getURLContent line is the equivalent of pointing your browser to that url. Because the page is XML, we then read or parse the page with xmlParse, creating an R object representation of all that XML. We are ready to rock and roll.
If you look closely at the XML, you’ll see all these
<dwc:Genus>Oreochromis</dwc:Genus>
[/xml]
Clearly these are telling us the family and the Genus for this creature. The computer just has to look for the
To grab this text using R, we simply ask for the value of the node named dwc:Family:
[source lang=”r”] Family <- xmlValue(getNodeSet(doc, "//dwc:Family")[[1]])Family
[/source]
There’s two commands combined in that line. getNodeSet() is the first, getting any nodes that have the name dwc:Family anywhere (the // means anywhere) in the document. The [[1]] indicates that we want the first node it finds (since there’s only one of these in the entire document). Specifying which node we want makes use of the xpath syntax, a powerful way of navigating XML which we’ll use more later.
The xmlValue command just extracts the contents of that node, now that we’ve found it. If we ask R for the value it got for Family, it says Cichlidae, just as expected.
That was easy. We can do the same for Genus, Scientific Name, etc. As we go down the XML document, we see some of the information we want isn’t given a uniquely labeled node.
For instance, the information for size, habitat, and distribution are all under nodes called
<dc:identifier>FB-Size-2</dc:identifier>
… other stuff ….
<dc:description> Text We need </dc:description>
… other stuff ….
</dataObject>
[/xml]
That identifier node tells us that this dataObject has size information. We can grab the content of that dc:description by first finding the identifier that says FB-Size-2, going up to it’s parent dataObject, and asking that dataObject for it’s child node called description. Like this:
[source lang=”r”] size_node <- getNodeSet(doc, paste("//dc:identifier[string(.) =FB-Size-", fish.id, "’]/..", sep=""))
size <- xmlValue( size_node[[1]][["description"]] )
[/source]
Okay, so that looks crazy complicated — guess we got in a bit deep. See that link to xpath above? That’s a wee tutorial that will explain most of what’s going on here. It’s pretty simple if you take it slow. Using these kind of tricks, we can extract just the information we need from the XML.
Meanwhile, if you want the fast-track option, I’ve put this together in a little R package called rfishbase. The package has a function called fishbase() which extracts various bits of information using these calls. Once you get the hang of it you can modify it pretty easily. I do a little extra processing to get numbers from text using Regular Expressions, a sorta desperate but powerful option when everything isn’t nicely labeled XML.
Using this function we query a fish id number and get back the data in a nice R list, which we can happily manipulate. Here’s a quick demo:
[source lang=”r”] require(rfishbase)## a general function to loop over all fish ids to get data
getData <- function(fish.ids){
suppressWarnings(
lapply(fish.ids, function(i) try(fishbase(i)))
)
}
# A function to extract the ages, handling missing values
get.ages <- function(fish.data){
sapply(fish.data, function(out){
if(!(is(out,"try-error")))
x <- out$size_values[["age"]]
else
x <- NA
x
})
}
# Process all the XML first, then extract the ages
fish.data <- getData(2:500)
yr <- get.ages(fish.data)
# Plot data
hist(yr, breaks=40, main="Age Distribution", xlab="age (years)");
[/source]
Note we take a bit of care to avoid the missing values using try() function. Here’s the results:
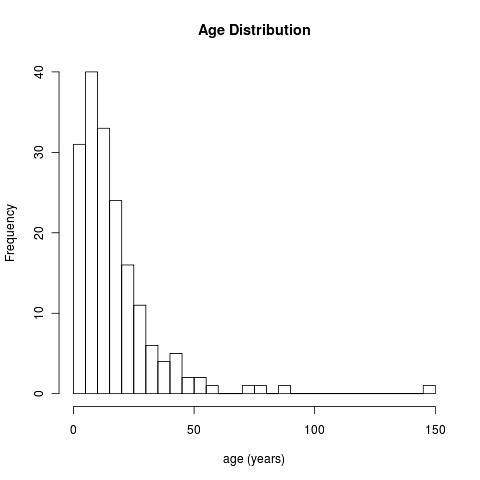
Kinda cool, isn’t it?


Recent Comments